How to run a job?
New alignment mode
Launch a new mapping between two fasta files and dot plot it.
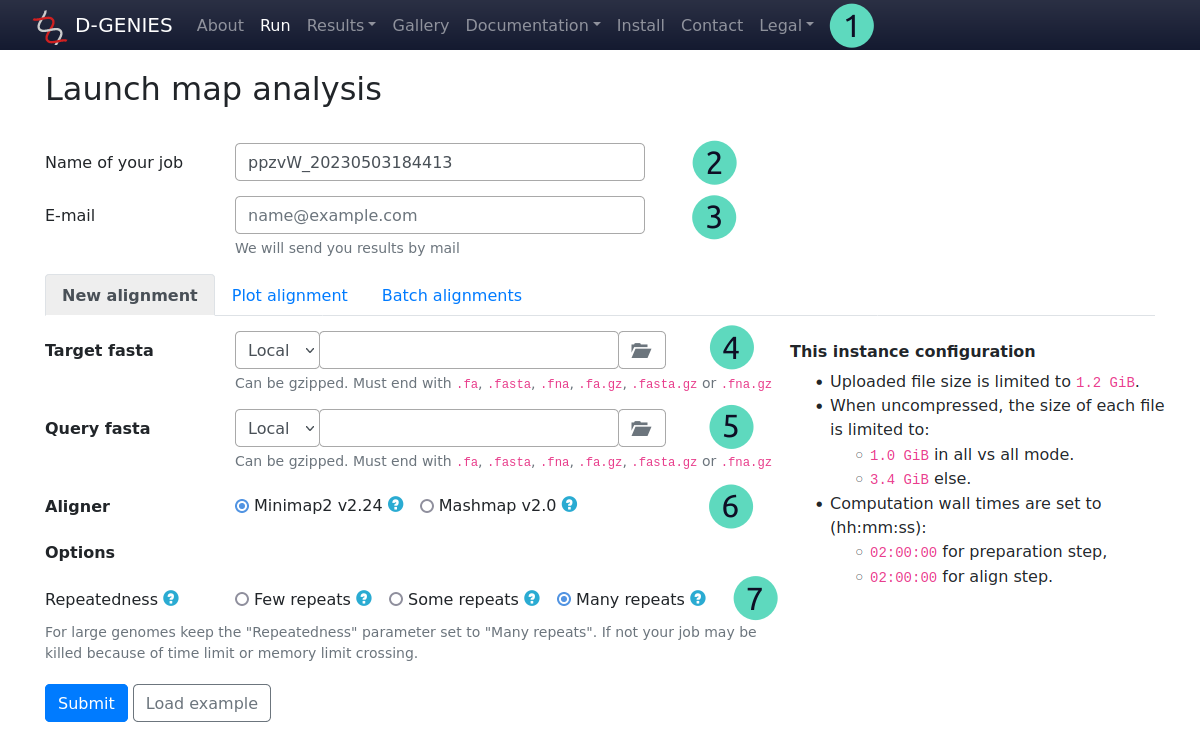
(1) Main menu
You just need to click on the main menu run tab, and fill the fields. All results will be stored in the result menu.
(2) Updatable job name
Required field
A unique job name is set automatically. You can change it. Note that if a job already exists with the same name, it will be automatically renamed.
(3) User email
Required field
Please enter your email. When the job is finished, you will receive a mai. Some features of the result page will also send you a mail to this address (see manual).
(4) Target fasta
Required field
With the selector at the left, you can choose to select a local file or enter an URL. For a local file, click on the button at the right to select it.
Files must be in fasta format. We recommend using gzipped files to preserve bandwidth and speed job submission.
Allowed extensions: fa, fasta, fna, fa.gz, fasta.gz, fna.gz
Max file size: 1 GiB (4 GiB once uncompressed, 3 GiB in all-vs-all mode)
(5) Query fasta
Optional field
Works like the target fasta. If not given, target file will be mapped to itself, in all-vs-all mode.
Max file size: 1 GiB (4 GiB once uncompressed)
(6) Aligner
You can choose aligner to use for mapping. By default, it's minimap2.
If your job fails due to memory limit reached, you can try mashmap. It uses less resources. But is only suitable for highly similar genomes as it only detect matches with more than 75% of identity.
(7) Aligner options
If the aligner as some options, you can choose some. For minimap2, we provide a repeatedness option which is related to number of repetitions found in sequences. By default, it is set to few repeats which is the current minimap2 default (-k 0.0002 in minimap2 parameters).
If your job fails due to memory limit or computation walltime reached, you can use some repeats (-k 0.002) or many repeats (-k 0.02) as an alternative to mashmap.
Plot alignment mode
Dot plot an existing alignment file.
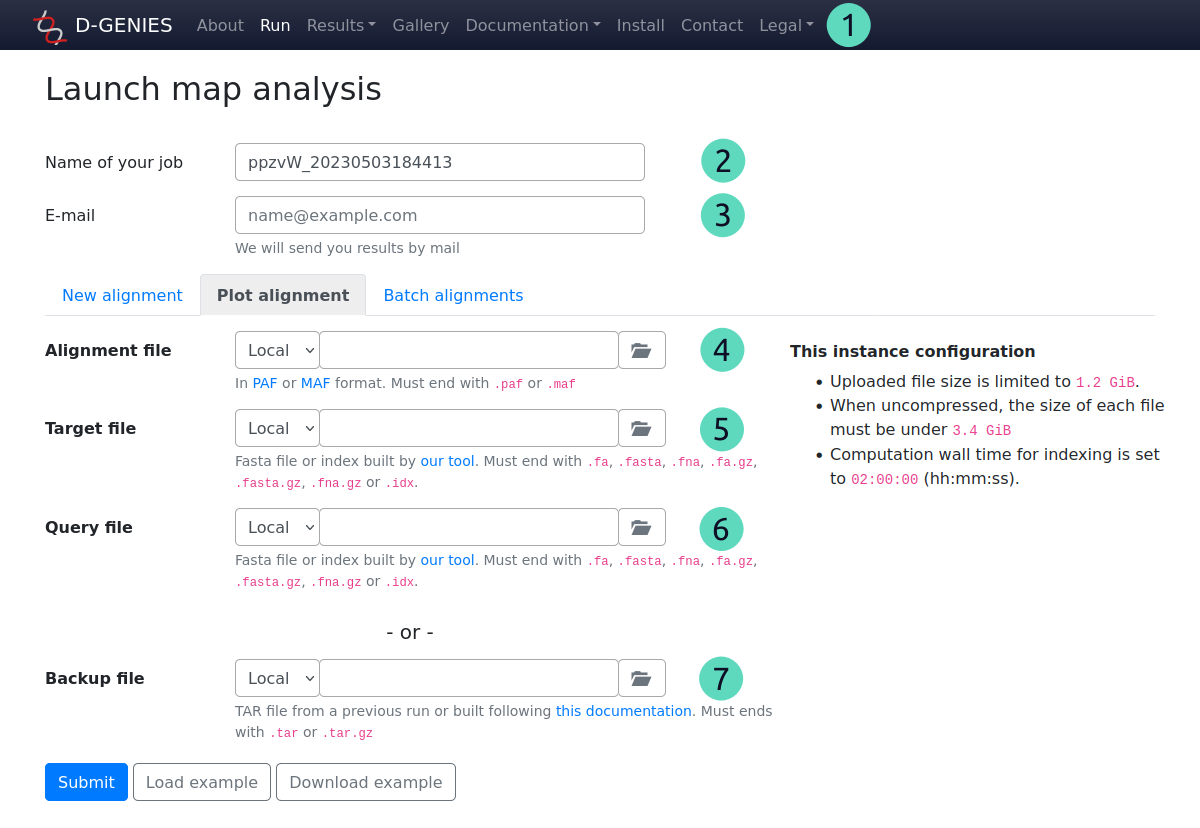
For numbers from 1 to 3, see previous section.
(4) Alignment file
Required field (except if backup file is filled, see bellow)
An alignment file in PAF or MAF format.
Allowed extensions: paf, maf
With the selector at the left, you can choose to select a local file or enter an URL. For a local file, click on the button at the right to select it.
(5) Target file
Required field (except if backup file is filled, see bellow)
Can be a fasta file or the corresponding index file.
To improve bandwidth and computation time, we recommend to use the index file. This file format is described here. You can use our tool to build it.
Allowed extensions:
Fasta: fa, fasta, fna, fa.gz, fasta.gz, fna.gz
Index: idx
With the selector at the left, you can choose to select a local file or enter an URL. For a local file, click on the button at the right to select it.
(6) Query file
Optional field
Can be the fasta file or the corresponding index file.
Works like the target file.
(7) Backup file
Optional field
If you downloaded the backup file from a previous job, you can enter it here to restore the dot plot. In this case, don't fill previous fields, only this one is required.
With the selector at the left, you can choose to select a local file or enter an URL. For a local file, click on the button at the right to select it.
Batch mode
Launch a batch of jobs, composed of a new alignments and plots.
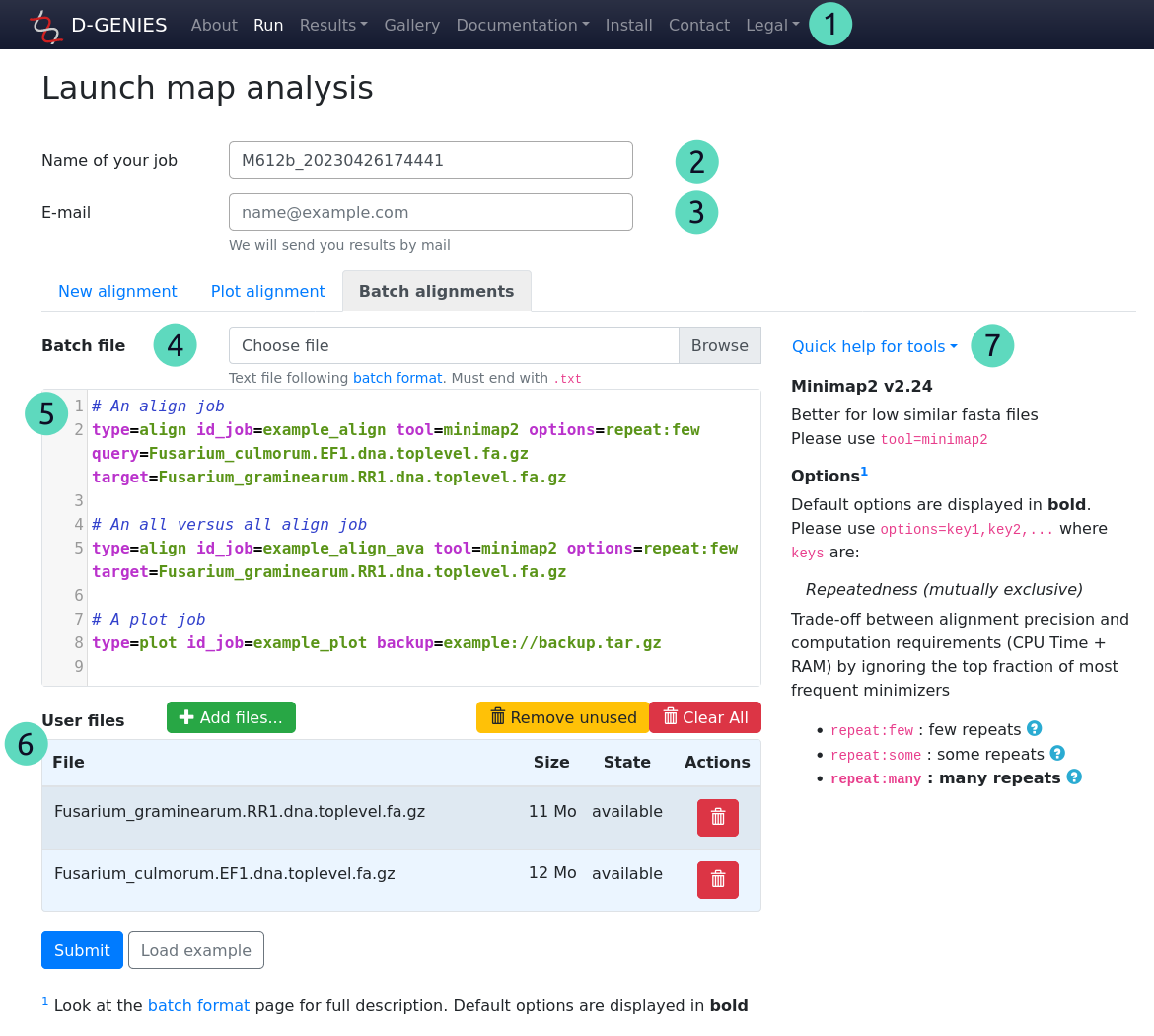
For numbers from 1 to 3, see previous section.
(4) Batch file
Optional field
Load a text file that will populate the text box field under. Syntax describing jobs can be found at batch format description page.
(5) Batch jobs
Required field
Batch jobs described in as text. Expected syntax can be found at batch format description page.
It can be populate by loading a file in Batch file field, but also by copy-pasting text directly or drag-dropping text file on it.
Some text editor shortcuts, like CTRL+Z for undo or CTRL+SHIT+Z for redo, can be use in this text box.
(6) User files
Optional field, depending of Batch jobs field content.
Local files must be added here either by clicking on Add files... button or by drag-dropping files or directories on file listing.
Each files can be removed individually by clicking on its trash button in the listing.
Unused files can be directly removed from listing by clicking on Remove unused and all files can be removed by clicking on Clear All button.
(7) Quick help for tools
Not an input field
This menu describes the syntax expected for tools and their options .